



Abstract
Acetylcholine (ACh) plays a permissive role in developmental plasticity of fibers from the lateral geniculate nucleus (LGN) to the primary visual cortex (V1). These fibers remain plastic and express long-term potentiation (LTP) in adult rodents, but it is not known if ACh modulates this form of plasticity in the mature V1. We show that, in anesthetized rats, theta burst stimulation (TBS) of the LGN using 5 or 40 theta cycles produced moderate (∼20%) and stronger (∼40%) potentiation, respectively, of field postsynaptic potentials recorded in the ipsilateral V1. Basal forebrain stimulation (100 Hz) 5 min after TBS enhanced LTP induced by both weak (5 theta cycles) and strong (40 theta cycles) induction protocols. Both effects were reduced by systemic administration of the muscarinic receptor antagonist scopolamine. Basal forebrain stimulation did not enhance LTP when applied 30 min after or 5 min prior to TBS, suggesting that ACh affects early LTP induction mechanisms. Application of the cholinergic agonist carbachol in V1 by means of reverse microdialysis mimicked the effect of basal forebrain stimulation. We conclude that heterosynaptic facilitation of V1 plasticity by ACh extends beyond early postnatal maturation periods and acts to convert weak potentiation into pronounced, long-lasting increases in synaptic strength.
Introduction
Classic studies by Hubel and Wiesel first demonstrated that visual experience plays a pivotal role in the development of the primary visual cortex (V1). Reducing visual input by means of monocular deprivation during early postnatal life leads to a loss of visual responsiveness of V1 neurons to inputs from the deprived eye (Wiesel and Hubel 1963; Shatz and Stryker 1978; Wiesel 1982). Initial studies suggested that this ocular dominance plasticity is restricted to an early postnatal period (Hubel and Wiesel 1970; Olson and Freeman 1980; Katz and Crowley 2002). Surprisingly, recent experiments demonstrate that, at least for rodents, similar effects of monocular deprivation are also seen in the mature cortex of adult animals (Sawtell and others 2003; Pham and others 2004; Tagawa and others 2005). The hypothesis that the mature V1 retains a significant potential to express activity-dependent plasticity is further supported by evidence that thalamic inputs to V1 readily express long-term potentiation (LTP) and long-term depression (LTD) in adult rats in vivo (Rittenhouse and others 1999; Heynen and Bear 2001). The parallels among the molecular mechanisms supporting ocular dominance plasticity and LTP/LTD suggest that mechanisms recruited during experimental LTP/LTD induction may also be responsible for plasticity resulting from altered visual input (Heynen and Bear 2001; Heynen and others 2003; Sawtell and others 2003; Lu and Constantine-Paton 2004; Malenka and Bear 2004).
Cholinergic inputs to sensory cortex are known to play important roles in several types of activity-dependent synaptic modifications (Metherate and others 1987; Edeline 1999; Rasmusson 2000; Verdier and Dykes 2001; Weinberger 2004). Axons originating in the basal forebrain provide the cholinergic innervation of V1 (Bigl and others 1982; Luiten and others 1987), and cholinergic terminals are particularly dense in layers II/III and V of the rat V1 (Gu 2003). Infusions of muscarinic receptor antagonists into V1 inhibit ocular dominance plasticity following monocular deprivation of kittens (Gu and Singer 1993). Further, orientation tuning of V1 neurons can be shifted when presentations of suboptimal orientation stimuli are paired with iontophoretic application of acetylcholine (ACh; Greuel and others 1988). Finally, in vitro studies have shown that ACh or muscarinic receptor activation facilitates the induction of both LTP and LTD in slices of V1, effects that appear to be mediated by an enhancement of N-methyl-D-aspartate (NMDA) receptor conductances (Bröcher and others 1992; Kirkwood and others 1999; Kojic and others 2001; Gu 2003). To date, experiments examining a cholinergic modulation of LTP induction in V1 in vivo have not been carried out.
Here, we demonstrate that, during a well-defined time window following induction, endogenous ACh facilitates thalamocortical LTP in V1 in vivo by increasing the magnitude of potentiation and converting transient potentiation into stable LTP. Similar effects are seen with cholinergic agonist application locally in V1. Thus, in the mature visual cortex, heterosynaptic mechanisms involving ACh continue to promote plasticity by facilitating the ability of thalamic signals to initiate molecular cascades underlying long-lasting increases in synaptic strength.
Materials and Methods
Subjects and Surgical Preparation
All experiments were conducted in accordance with published guidelines of the Canadian Council on Animal Care and approved by the Queen's University Animal Care Committee. The experiments were conducted on adult (300–450 g), male Long-Evans rats housed in a colony room (12/12-h light cycle) with free access to food and water. Each rat was used for only one experiment (i.e., received only one stimulation protocol).
Experimental procedures were carried out under deep urethane anesthesia (1.5 g/kg, intraperitoneal [i.p.], administered in three 0.5 g/kg doses every 20 min and supplemented as necessary). Urethane supplements (typically in the range of an additional 0.5 g/kg, given prior to the onset of data collection) were sufficient to ensure that spontaneous high-frequency, low-amplitude activation in the electrocorticogram (ECoG, see below) did not occur during the course of the experiment. Rats were placed in a stereotaxic apparatus and body temperature was maintained between 36 and 37 °C by means of an electrical heating blanket. The skull was exposed, and small skull holes (all ipsilateral) were drilled to allow access to the basal forebrain (anterior-posterior (AP) −1.0 to −1.4, lateral (L) +2.5, ventral (V) −8.0), lateral geniculate nucleus (LGN; AP −3.8 to −4.0, L +3.8 to +4.0, V −4.5 to −5.0), and V1 (AP −7.5, L +3.5, V < −0.5). Final placements of LGN stimulation and V1 recording electrodes were adjusted to elicit optimal field postsynaptic potential (fPSP) amplitude and augmenting responses (at 100-ms interpulse intervals) in VI in response to LGN stimulation. In a subset of experiments, recordings of multiunit activity in the LGN through the stimulation electrode were performed to confirm visually (flash) evoked neural responses at the sites used for stimulation of inputs to V1.
Electrophysiology
Stimulation of the LGN (0.2-ms negative pulses every 30 s, intensity adjusted to yield 50–60% of maximal fPSP amplitude) was provided by a concentric bipolar electrode (Rhodes Medical Instruments Series 100, David Kopf, Tujunga, CA) connected to a stimulus isolation unit providing a constant current output (PowerLab/16 s system with ML 180 Stimulus Isolator, ADInstruments, Toronto, Canada). For LTP induction, theta burst stimulation (TBS) of the LGN was applied in the form of bursts (5 pulses at 100 Hz, intensity and duration as for single-pulse stimulation) that were repeated (2–40) at 5 Hz. For stimulation regimes consisting of 20–40 theta bursts, 10 consecutive bursts (at 5 Hz) were applied, and this was repeated every 10 s until the total number of theta bursts had been delivered. Basal forebrain stimulation was applied through a monopolar electrode (Teflon insulated stainless steel, 125-μm tip diameter) delivering 0.5 mA, negative pulses (0.2-ms duration) at 100 Hz for 0.5 s × 10 every 10 s.
The fPSPs in V1 was differentially recorded (Teflon insulated stainless steel wire, 125-μm tip diameter) against a ground screw placed in the bone overlying the cerebellum. The signal was amplified, digitized (10 kHz), and stored for subsequent offline analysis using the PowerLab system. In addition, ECoG activity was recorded through the V1 electrode to assess the effect of basal forebrain stimulation. The cortical signal was amplified, digitized (200 Hz), filtered (0.3–40 Hz), and analyzed offline by means of spectral analysis (PowerLab running Chart software, v. 3.6.5).
Experimental Procedures
For each experiment, 60 initial baseline fPSPs were recorded every 30 s, followed by TBS of the LGN. Basal forebrain stimulation was applied 5 or 30 min after TBS or 5 min prior to TBS. Recordings of fPSP (every 30 s) continued for 2–4 h following TBS. For experiments assessing the effects of muscarinic and nicotinic cholinergic receptor blockade, scopolamine (5 mg/kg, i.p.) or scopolamine + mecamylamine (5 mg/kg, i.p.) was administered 30 min prior to the commencement of baseline fPSP recordings. For experiments employing local, intracortical carbachol application, the cortical recording electrode was taped to a microdialysis probe (Mab 6 probe, 2-mm polyether sulfone membrane, 15 000 Dalton cutoff, 0.6-mm diameter, S.P.E. Limited, North York, Ontario, Canada). The dialysis membrane was located immediately adjacent to the recording electrode and extended 1 mm beyond the electrode tip. The dialysis probe electrode assembly was connected to a 1-mL syringe driven by a dialysis pump (Univentor 801, S.P.E. Limited) and continuously perfused with artificial cerebrospinal fluid (aCSF, 1 μL/min). Carbachol (0.01–10 mM) was added to the aCSF and applied for 15 min immediately following TBS. All chemicals were obtained from Sigma/Research Biochemicals, Oakville, Ontario, Canada.
At the end of the experiment, rats were perfused through the heart with 10% formalin, their brains were removed, and standard histological techniques were used to verify all electrode placements. Data obtained with inaccurate placements were excluded from the data analyses.
Data Analysis
The maximum amplitude of the fPSP was computed offline. Amplitude data were averaged over 10-min intervals, and these averages were normalized by dividing all data for one rat by the average baseline (pre-TBS) amplitude of that animal. For ECoG activity, spectral analyses (0.2-Hz bins) for one 10-s epoch recorded immediately prior to and one 10-s epoch recorded immediately after basal forebrain stimulation were computed using Chart (v. 3.6.5) software. All data are expressed as mean ± standard error of mean. For statistical analyses, analysis of variance (ANOVA) and, where statistically appropriate, simple effects tests were computed using the software package CLR Anova (v. 1.1, Clear Lake Research Inc., Houston, TX).
Results
Characteristics of fPSPs and Determination of LTP Induction Threshold
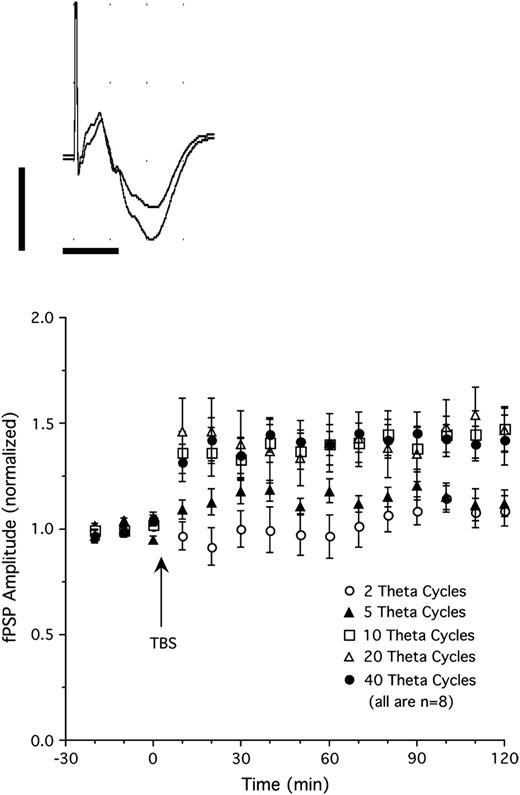
Single-pulse stimulation of the LGN elicited negative-going fPSP in the superficial layers (I–II) of V1 with a peak latency of about 11–14 ms (Fig. 1 top). Heynen and Bear (2001) showed that this potential reflects a current sink in the upper (mostly II–III) cortical layers of V1 in the rat. We determined the threshold for LTP induction by applying different TBS regimes consisting of 2–40 theta cycles (5 pulses at 100 per cycle, repeated at 5 Hz). Thalamic TBS with 2 theta cycles (n = 8, Fig. 1 bottom) failed to induce LTP in V1 (fPSP amplitude 102% of baseline when averaged over 2 h following TBS; P = 0.28). Stimulation with 5 theta cycles (n = 8; Fig. 1 bottom) induced moderate potentiation that approached statistical significance (114% of baseline over 2 h after TBS; P = 0.07). Application of 10, 20, or 40 theta cycles (n = 8 per group; Fig. 1 bottom) all produced clear LTP with similar levels of potentiation over the 2 h following TBS (140%, 142%, and 141% of baseline, respectively; all P values < 0.01). Evoked potentials shown in Figure 1 (top) show an example of increased fPSP amplitude after 20 cycle TBS. For the subsequent experiments, TBS consisting of 5 and 40 theta cycles was used to produce, respectively, threshold and presumably maximal potentiation of thalamocortical transmission.
{kind=link}
Top: fPSPs recorded in V1 in response to single-pulse stimulation of the LGN before (smaller amplitude fPSP) and after thalamic TBS (20 theta cycles, larger amplitude fPSP). Each trace is an average of 20 individual sweeps (calibrations are 1 mV and 10 ms). Bottom: Changes in fPSP amplitude produced by different theta burst protocols. Stimulation using 2 theta cycles (5 pulses at 100 Hz, repeated at 5 Hz) failed to induce synaptic potentiation. Five theta cycle stimulation induced moderate potentiation, whereas stimulation consisting of 10, 20, or 40 theta cycles induced strong, stable potentiation of fPSP amplitude (n = 8 per group).
Basal Forebrain Stimulation Enhances Thalamocortical LTP
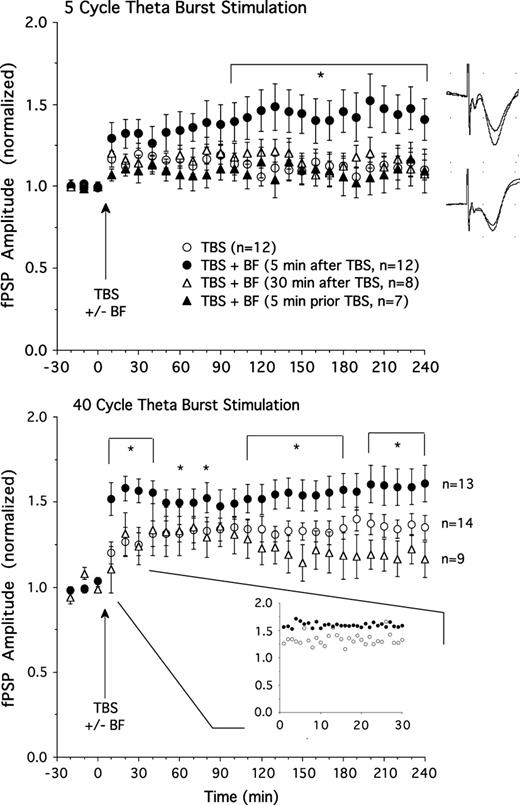
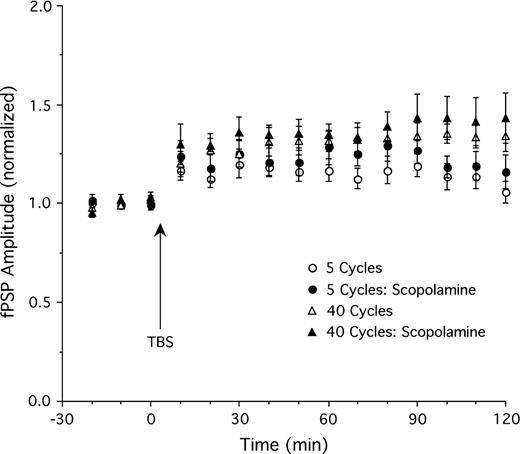
Consistent with the previous experiment, TBS consisting of 5 theta cycles (n = 12) induced moderate potentiation of fPSP amplitude (Fig. 2 top; maximal potentiation of ∼20% above baseline 30 min after TBS, 10% at the end of the experiment 4 h after TBS). Basal forebrain stimulation (10 × 0.5 s, 100 Hz, 0.5 mA; see Fig. 3 for electrode placements) produced an enhancement of LTP when it was applied 5 min after TBS (n = 12, F26,572 = 3.1, P < 0.01 for ANOVA interaction of stimulation group and time). Combined TBS and basal forebrain stimulation (Fig. 2 top) resulted in a maximal potentiation of ∼50% (41% at the end of the experiment 4 h after TBS). However, enhancement of LTP was not apparent when basal forebrain stimulation was delayed 30 min following thalamic TBS (n = 8; Fig. 2 top; F26,468 = 0.96, P = 0.52; 22% maximal potentiation, 8% at the end of the experiment) or when it occurred 5 min prior to TBS (n = 7; Fig. 2 top; F26,442 = 0.69, P = 0.88; 15% maximal potentiation, 9% at the end of the experiment). Thus, stimulation of the basal forebrain allows thalamic inputs to initiate long-lasting synaptic potentiation, but only during a fairly narrow time window following thalamic excitation.
{kind=link}
Top: Amplitude of the fPSP before and after thalamic TBS with 5 theta cycles. TBS alone (n = 12) produced moderate potentiation. Additional stimulation of the basal forebrain (BF) 5 min after (n = 12), but not 30 min (n = 8) after, or 5 min prior to (n = 7) TBS resulted in greater LTP relative to TBS alone. Examples of fPSPs (averages of 10 sweeps) showing LTP in a rat given 5 cycle TBS plus BF stimulation (top) and minimal potentiation in a rat receiving only 5 cycle TBS (bottom). Bottom: Thalamic TBS consisting of 40 theta cycles (n = 14) produced stable LTP which was further enhanced by BF stimulation applied 5 min (n = 13), but not 30 min (n = 9), after TBS. *P < 0.05 for simple effect comparisons of TBS only and TBS plus BF stimulation (5 min after TBS) groups. The insert depicts fPSP amplitude for the TBS plus BF and TBS only groups using smaller (1 min) time bins for the 30-min time window immediately following BF stimulation. Note that rats receiving BF stimulation showed an enhanced fPSP amplitude right from the onset of this 30-min sampling period.

{kind=link}
Placements of stimulation electrodes in the basal forebrain. Numbers indicate distance from bregma in millimeters. CPu, caudate putamen; LGP, lateral globus pallidus; ic, internal capsule; LH, lateral hypothalamus; SI, substantia innominata; VP, ventral pallidum. Adapted from the atlas by Paxinos and Watson (1998).
We examined whether the effect described above is limited to a moderate LTP induction protocol, or whether LTP induced by a stronger TBS protocol can be further enhanced by basal forebrain stimulation. Stimulation of the LGN using 40 theta cycles (n = 14; Fig. 2 bottom) produced clear LTP (∼40% maximal potentiation, 35% potentiation 4 h after TBS). Basal forebrain stimulation 5 min after TBS (n = 13; Fig. 2 bottom) resulted in a significant further enhancement of LTP (∼60% maximal potentiation; F26,650 = 2.9, P < 0.01 for ANOVA interaction of stimulation group and time). We examined whether basal forebrain–induced LTP facilitation occurred immediately after basal forebrain stimulation or more gradually by replotting data using smaller 1-min time bins (insert in Fig. 2 bottom). These data representing fPSP amplitude for the initial 30 min immediately following basal forebrain stimulation indicate that fPSP amplitude was already enhanced during the first minute and reached maximal levels of potentiation at about 4 min after basal forebrain stimulation.
As was the case for potentiation induced by 5 theta cycle TBS, basal forebrain stimulation did not enhance strong LTP when delivered 30 min following TBS (∼36% maximal potentiation; n = 9). In fact, it appeared as if 30-min-delayed basal forebrain stimulation depressed fPSP amplitude during the last 2 h of the experiment relative to animals receiving only TBS (Fig. 2 bottom; F26,546 = 1.6, P = 0.03).
Basal Forebrain–Induced LTP Enhancement Involves Cholinergic Muscarinic Receptor Activation
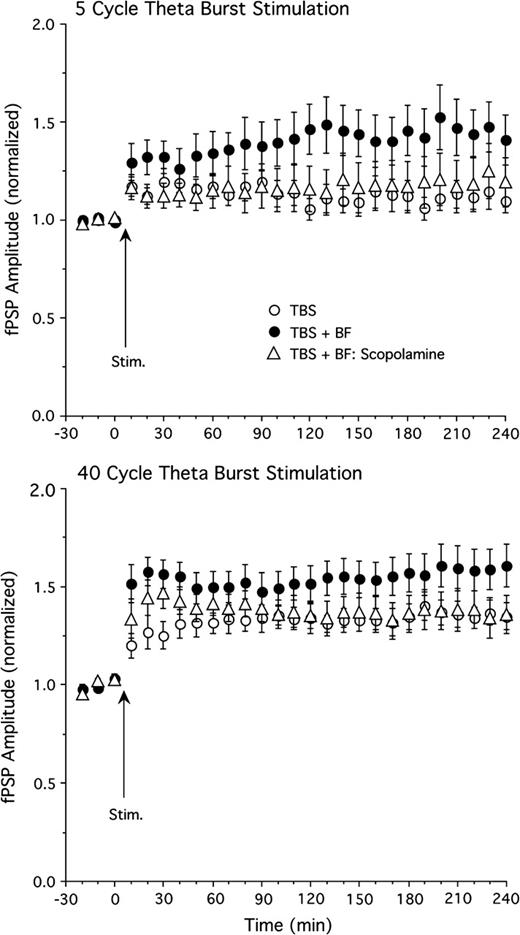
The dependence of basal forebrain–elicited LTP enhancement on cholinergic muscarinic mechanisms was assessed by administering scopolamine (5 mg/kg, i.p.) 30 min prior to the onset of the experiment. Scopolamine strongly reduced the effect of basal forebrain stimulation to enhance LTP. In the presence of scopolamine, basal forebrain stimulation (applied 5 min after TBS) no longer produced a significant enhancement of fPSP amplitude in rats receiving 5 cycle TBS (Fig. 4 top, n = 11, F26,546 = 0.8, P = 0.75 for ANOVA interaction of group and time; 26% maximal potentiation). However, in several experiments, some residual basal forebrain enhancement was apparent during the last hour of the experiment (Fig. 4 top), suggestive of a small nonmuscarinic component in basal forebrain–induced LTP enhancement. We tested if nicotinic receptors are responsible for this small, scopolamine-resistant enhancement by treating a group of rats (n = 6) with scopolamine and the nicotinic receptor antagonist mecamylamine (5 mg/kg, i.p., 30 min prior to onset of experiment). However, even combined scopolamine–mecamylamine treatment did not abolish the residual enhancement of LTP late in the experiment (fPSP potentiation averaged during the last 30 min of experiment was 12% for TBS only, 44% for TBS and basal forebrain stimulation, 23% for TBS and basal forebrain stimulation in the presence of scopolamine, and 28% for TBS and basal forebrain stimulation in the presence of scopolamine and mecamylamine). Thus, nicotinic receptor mechanisms do not appear to contribute to this effect.
{kind=link}
The effect of scopolamine (5 mg/kg, i.p.) on LTP enhancement induced by basal forebrain (BF) stimulation. The BF-induced enhancement of LTP induced by 5 cycle TBS (top) and 40 cycle TBS (bottom) was reversed by scopolamine (n = 11 and 9 for scopolamine groups; other groups identical to those in Fig. 2).
Scopolamine exerted a similar effect when LTP was induced by 40 cycle TBS (n = 9; Fig. 4 bottom), with basal forebrain stimulation no longer producing a significant enhancement of LTP (F26,546 = 1.2, P = 0.28; 47% maximal potentiation). Again, however, there appeared to be some residual basal forebrain enhancement in the presence of scopolamine, which, in this case, was apparent during the first 30 min following LTP induction.
Control experiments (Fig. 5) showed that, in the absence of basal forebrain stimulation, scopolamine did not reduce LTP induced by either 5 cycle TBS (n = 8, 29% maximal potentiation; comparison of TBS vs. TBS scopolamine groups, F1,18 = 0.9, P = 0.35 and F14,252 = 0.6, P = 0.87 for group effect and group by time interaction, respectively) or 40 cycle TBS (n = 5, 43% maximal potentiation; F1,17 = 0.2, P = 0.65 and F26,442 = 0.5, P = 0.98 for group effect and group by time interaction, respectively). Thus, a nonspecific suppression of thalamocortical LTP does not appear to contribute to the effect of scopolamine to reduce basal forebrain–mediated LTP enhancement.
{kind=link}
The effect of scopolamine (5 mg/kg, i.p.) on LTP in the absence of basal forebrain stimulation. Scopolamine had no effect on LTP induced by either 5 (n = 8) or 40 cycle (n = 5) TBS (other groups as in Fig. 2).
Effects of Basal Forebrain Stimulation on Nonpotentiated Thalamocortical fPSP and ECoG
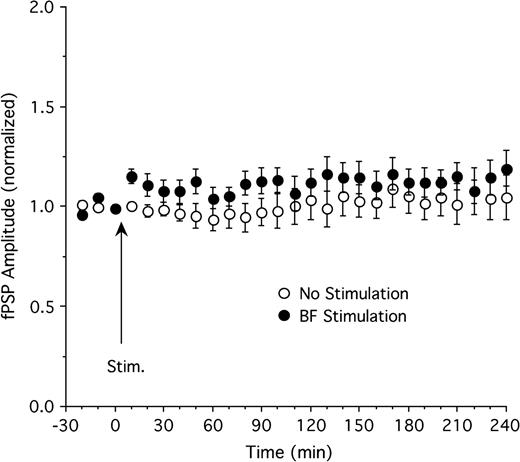
The effect of basal forebrain stimulation on thalamocortical fPSP in the absence of thalamic TBS was evaluated in a separate set of experiments. In control animals receiving test pulses in the LGN but no basal forebrain stimulation (n = 11), fPSP amplitude remained relatively stable throughout the experiment, ranging from 94% to 109% of baseline amplitude (Fig. 6). There was a slight increase in fPSP amplitude following basal forebrain stimulation (n = 11), with amplitude in the range of 104–119% of baseline during the 4 h following stimulation (Fig. 6). However, this effect did not reach statistical significance (F26,520 = 0.7, P = 0.87 for ANOVA interaction of group and time; main effect of group also not significant, P = 0.22).
{kind=link}
The effect of basal forebrain (BF) stimulation on fPSP amplitude in the absence of thalamic TBS. BF stimulation (n = 11) resulted in a transient, statistically insignificant increase of fPSP amplitude relative to control animals (n = 11).
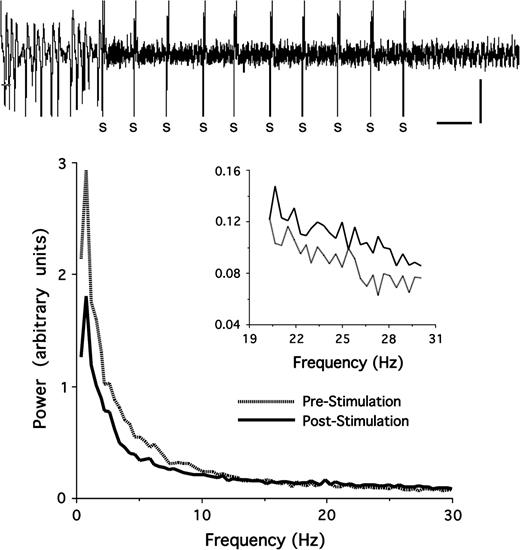
Analysis of the ECoG recorded in V1 (n = 23) prior to basal forebrain stimulation (10-s epochs) revealed that, under deep urethane anesthesia, the ECoG was dominated by large-amplitude (up to 1 mV) slow oscillations with peak power in the delta (0.5–3.9 Hz) frequency band (Fig. 7). Stimulation of the basal forebrain reduced slow delta activity, which, in the majority of cases, was replaced by lower amplitude (<0.5 mV) activity (Fig. 7). Power spectral analyses showed that power in the delta, theta (4–7.9 Hz), and low-alpha bands (8–10 Hz) was suppressed even though peak power still occurred in the delta band (Fig. 7). Higher frequency beta (20–30 Hz) power was increased following basal forebrain stimulation (Fig. 7 inset). The effect of basal forebrain stimulation on the ECoG lasted for less than 60 s.
{kind=link}
The effect of basal forebrain (BF) stimulation on ECoG activity in V1. Top: Stimulation of the BF (indicated by S) suppressed slow, large-amplitude oscillations present in urethane anesthetized rats prior to the stimulation (calibration: 10 s, 0.5 mV). Bottom: Power spectral analyses for 10-s epochs recorded before and (Pre-Stimulation) and after (Post-Stimulation) BF stimulation. Stimulation suppressed power in the lower frequency bands, particularly delta (0.5–3.9 Hz) and theta (4–7.9 Hz) power, whereas higher frequency beta power (20–30 Hz, shown in the insert) was enhanced following BF stimulation. Power spectra shown are averages of 23 rats and are based on 10 s of continuous ECoG activity recorded immediately prior to the first and immediately after the last of 10 stimulation trains applied to the BF.
Correlational analyses were performed to assess if the degree of delta suppression (the most sensitive index of basal forebrain–induced ECoG changes, measured during the initial 10 s following stimulation) or beta enhancement predicted the strength of LTP observed at the end of the experiment. For experiments using 5 theta cycle TBS, there was no relation between the degree of basal forebrain–induced delta suppression and final LTP strength (r = 0.15, P = 0.63, n = 12, Spearman rank order correlation between delta suppression and fPSP amplitude 4 h after TBS). For experiments using 40 theta cycle stimulation, this correlation approached significance (r = 0.55, P = 0.08, n = 11). The correlations between beta enhancement and final LTP strength also did not reach statistical significance (5 cycle TBS, r = 0.46, P = 0.19; 40 cycle TBS, r = 0.25, P = 0.47).
Carbachol Application in V1 Enhances Thalamocortical LTP
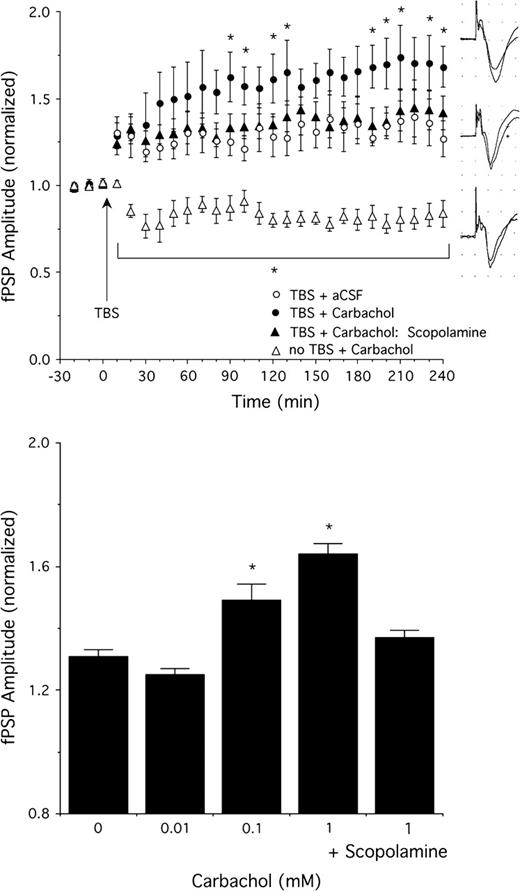
We assessed if direct application of the cholinomimetic carbachol in V1 by means of reverse microdialysis mimicked the effects of basal forebrain stimulation to enhance thalamocortical LTP. In these experiments, 5 cycle TBS alone (in the presence of continuous aCSF perfusion) resulted in a ∼30% enhancement of fPSP amplitude averaged over the 4 h following induction (40% maximal potentiation, n = 8, Fig. 8). Carbachol (concentration range between 0.01 and 1 mM, n = 4 for 0.01 mM, n = 8 for 0.1 and 1 mM) applied for 15 min immediately after TBS resulted in a concentration-dependent enhancement of LTP, with a maximal effect seen for 1 mM (Fig. 8; 74% maximal potentiation; F78,624 = 1.4, P = 0.02 for ANOVA interaction of carbachol concentration and time; interactions also significant for individual comparisons of 0 vs. 0.1 and 1 mM carbachol, with carbachol-enhancing LTP relative to animals receiving aCSF, P = 0.0001 and 0.006, respectively). At 10 mM, carbachol produced strong depression of fPSP amplitude and failed to enhance LTP (not shown). Consistent with the effects of muscarinic blockade summarized above, the LTP enhancement produced by the optimal carbachol concentration (1 mM) was blocked by scopolamine treatment (Fig. 8; 45% maximal potentiation; scopolamine dose was 5 mg/kg, i.p., administered 30 min prior to the onset of baseline recordings, n = 6; comparison of 0 mM and 1 mM carbachol + scopolamine, F26,312 = 0.4, P = 0.9 for interaction of drug and time, group comparison also not significant, P = 0.7). In the absence of thalamic TBS, carbachol application for 15 min at the optimal concentration of 1 mM (n = 6) resulted in a suppression of fPSP amplitude to about 80% of baseline levels that lasted until the end of the experiment. Similar inhibitory effects in sensory cortex following muscarinic receptor activation have been described and likely reflect presynaptic inhibition (Kimura and others 1999), even though postsynaptic contributions have also been suggested (Oldford and Castro-Alamancos 2003).
{kind=link}
The effect of cortical carbachol application on fPSP amplitude with and without 5 cycle TBS. Top: Potentiation of fPSP amplitude by TBS was enhanced by carbachol (1 mM, n = 8) relative to animals receiving continuous aCSF application (n = 8). Pretreatment with scopolamine (5 mg/kg, i.p., n = 6) blocked the carbachol-induced LTP enhancement. Carbachol suppressed fPSP amplitude in animals that did not receive thalamic TBS (n = 6). Examples of fPSPs (averages of 10 sweeps each before and after TBS) in a rat given 5 cycle TBS plus carbachol (1 mM, top), only TBS (middle), and carbachol (1 mM) without thalamic TBS (bottom). Bottom: Concentration response data (n = 8 except n = 4 for 0.01 mM) for the effect of carbachol to enhance thalamocortical LTP. Scopolamine (5 mg/kg, i.p., n = 6) blocked the carbachol (1 mM)-induced LTP facilitation. Data shown are the average fPSP amplitudes between 2 and 4 h after TBS. *P < 0.05 relative to 0 mM carbachol.
Discussion
The present experiments demonstrate that cholinergic activity strongly enhances plasticity of thalamocortical inputs to V1 in adult rats. Previous work has shown that stimulation of the basal forebrain results in the release of ACh in the neocortex (Casamenti and others 1986; Metherate and others 1992). Our data show that basal forebrain stimulation facilitates moderate and strong thalamocortical LTP induced by TBS with 5 and 40 theta cycles, respectively. These effects were antagonized by scopolamine and occurred only when basal forebrain stimulation occurred in close temporal proximity (5 min) following thalamic TBS. Cortical carbachol application mimicked the effects of endogenous ACh release, suggesting that a local effect in V1 is sufficient to produce the enhancement of thalamocortical plasticity. Previous work has suggested that the type of LTP studied in our experiments results from the potentiation of monosynaptic thalamocortical synapses in cortical layer IV, as well as potentiation of intracortical synapses between layers IV and II–III (Heynen and Bear 2001). The functional consequence of LTP induction at these synapses is an enhanced cortical response to visual stimuli (Heynen and Bear 2001). Thus, our results suggest that ACh has the potential to facilitate long-lasting changes in the response of widespread networks in the mature V1 to visual inputs. Further, the mechanisms described here may well play a role in mediating facilitation of plasticity in other sensory areas (e.g., auditory, somatosensory) where ACh is known to promote activity-dependent plasticity (e.g., Metherate and others 1987; Edeline 1999; Rasmusson 2000; Verdier and Dykes 2001; McLin and others 2002a; Weinberger 2004).
The role of ACh in developmental plasticity of V1 in vivo is well established, in particular with regard to ACh as a permissive factor in the development of ocular dominance plasticity (Bear and Singer 1986; Gu and Singer 1993; Gu 2003). Further, in vitro studies have demonstrated that cholinergic receptor activation lowers the threshold for the successful induction of both LTP and LTD in slices of V1, effects involving M1 receptors, and alterations in NMDA receptor currents in response to afferent stimulation (Bröcher and others 1992; Kirkwood and others 1999; Kojic and others 2001). Similar effects of heterosynaptic LTP facilitation by ACh have been observed in the hippocampal formation in vitro (Huerta and Lisman 1993; Auerbach and Segal 1994) and in vivo (Leung and others 2003; Ovsepian and others 2004). There are several parallels between ocular dominance plasticity and NMDA-dependent LTP/LTD. Monocular deprivation in rodents leads to LTP-like, NMDA receptor–dependent enhancement of inputs from the nondeprived eye and concurrent LTD-like depression of inputs from the deprived eye (Rittenhouse and others 1999; Heynen and others 2003; Sawtell and others 2003). Thus, it is probable that LTP/LTD-like mechanisms are involved in mediating the activity-dependent refinement of V1 (Sawtell and others 2003; Lu and Constantine-Paton 2004; Malenka and Bear 2004).
The data presented here demonstrate that the role of ACh to regulate large-scale plasticity of visual cortex afferents in vivo is not limited to changes produced by prolonged alterations in visual input during early development. Facilitatory effects of endogenous ACh or exogenous cholinergic agonists on nonpotentiated activity in V1 have been reported (Sato and others 1987; Golmayo and others 2003), some of which are short lasting (50–100 ms; Lewandowski and others 1993) and could mediate cholinergic actions on modality-specific sensory enhancement and attentional processes (Sarter and Bruno 2000; McGaughy and others 2002; Fournier, Semba, and Rasmusson 2004). The long-lasting effects following basal forebrain stimulation observed here were strongly reduced by scopolamine treatment, indicative of a critical role of muscarinic receptor activation. Thus, it appears that noncholinergic basal forebrain efferents (γ-aminobutyric acidergic [GABAergic], peptidergic, and possibly glutamatergic; see Semba 2000) alone do not significantly contribute to basal forebrain–induced LTP facilitation. Scopolamine had no effect on LTP in the absence of basal forebrain stimulation, presumably due to insufficient levels of spontaneous ACh release under deep anesthesia. In the hippocampus in vivo, the modulatory effect of ACh on LTP is state specific, that is, a facilitatory effect of ACh can be demonstrated when LTP is induced during walking (high-ACh release) but not immobility (lower ACh release; Leung and others 2003). Together, these results indicate that, rather than acting as a general permissive agent, ACh selectively enhances plasticity during specific behavioral and/or cognitive states.
The present experiments indicate that ACh exerts its effects on thalamocortical LTP by means of muscarinic receptor activation, results that are consistent with other work on cholinergic influences on sensory cortex plasticity (see reviews by Rasmusson 2000; Gu 2003). The role of nicotinic receptors has not received much attention, especially with regard to the effects of endogenous ACh release rather than administration of an exogenous agonist. In our experiments, the effect of basal forebrain stimulation to enhance LTP was blocked by scopolamine, suggesting that nicotinic receptor activation alone is not able to produce this effect. Further, the small residual LTP enhancement that was present after scopolamine treatment was not abolished by additional mecamylamine administration, indicating that nicotinic receptors do not mediate this effect. However, in preliminary in vivo experiments in hippocampus (D. Habib and H.C. Dringenberg, unpublished data), we have observed that a mix of muscarinic and nicotinic mechanisms can be recruited by induction protocols consisting of pairing single-pulse stimulations of the septum and intrahippocampal glutamatergic fibers. These findings, if confirmed and extended to the neocortex, suggest that the precise patterns of cellular activity (e.g., spiking vs. bursting) in the basal forebrain may be able to recruit different patterns of receptor mechanisms to influence strength of cortical synapses.
It is worthwhile mentioning that the rat cerebral cortex contains a small percentage (1–2%) of bipolar cells that can contain choline acetyltransferase, mostly located in cortical layers II and III (Semba and Fibiger 1989). Current evidence suggests that these cortical cholinergic interneurons are involved in cerebrovascular regulation (Chédotal and others 1994), but it remains to be examined to what extent they are activated by basal forebrain inputs. Nevertheless, the presence of these cells in the rat cortex raises the possibility that ACh released from intracortical neurons contributes to effects such as those observed in the present experiments.
The present experiments are in agreement with work on cholinergic–glutamatergic interactions in several forebrain systems. It is known that ACh enhances NMDA-mediated synaptic responses and LTP in the striatum, hippocampus, and neocortex (Markram and Segal 1990; Huerta and Lisman 1993; Auerbach and Segal 1994; Calabresi and others 1998; Verdier and Dykes 2001; Leung and others 2003; Ovsepian and others 2004). Coactivation of NMDA and muscarinic receptors can synergistically interact to produce de novo protein synthesis in dendrites of hippocampal CA1 pyramidal cells (Feig and Lipton 1993). Protein synthesis is necessary for the induction of stable (late phase) LTP (Krug and others 1984; Frey and others 1988). Thus, LTP facilitation by the basal forebrain likely relates to a more efficient induction of protein synthesis and the resulting structural modifications of V1 synapses.
Interestingly, in slices of V1, the induction of LTP is age dependent, with reduced or no LTP observed in slices from mature animals. In the rat, stimulation protocols that reliably induce LTP in vitro in slices of 2- and 3-week-old animals fail to induce LTP at 4 weeks of age (Kato and others 1991). Similarly, V1 slices obtained from 60- to 80-day-old kittens are resistant to tetanic LTP induction, even though this effect can be overcome by additional muscarinic receptor stimulation (Kojic and others 2001). In contrast, thalamocortical LTP can be readily induced in V1 of adult animals in vivo (Heynen and Bear 2001; Dringenberg and others 2004; present results). Thus, like other sensory cortices (Edeline 1999; Weinberger 2004), V1 exhibits a potential for plasticity that extends beyond early developmental periods. The strength of potentiation does, however, depend on the neuromodulatory environment. As we show, increasing extracellular ACh lowers input requirements for successful induction of LTP so that threshold induction protocols produce robust potentiation when coupled with cholinergic enhancement. It is possible that the transmitter deficient environment typical of in vitro preparations contributes to the apparent limits on LTP in slices from older animals (limits that can be reduced by cholinergic receptor activation; Kojic and others 2001). Alternatively, the fact that large regions of V1 can be concurrently activated by thalamic stimulation may also account for the relative ease to obtain LTP in in vivo preparations (see Heynen and Bear 2001).
Recently, Hirata and Castro-Alamancos (2006) have proposed the intriguing hypothesis that neocortical synaptic enhancement induced by TBS of thalamocortical fibers is induced by a disinhibition of thalamocortical transmission, rather than a postsynaptic (cortical) modification of synaptic efficacy. These authors observed that thalamic inactivation abolished TBS-induced LTP in the barrel cortex in vivo, suggesting that intracortical mechanisms are insufficient to initiate LTP. For V1, however, there is strong evidence that intracortical synapses are potentiated after high-frequency fiber activation. For example, TBS applied to layers IV or II–III results in LTP in layers II–III and V–VI, respectively, in slices of V1, that is, in the absence of potential thalamic influences (Wang and Daw 2003; see also Heynen and Bear 2001). Further, we have observed that LTP elicited in the primary auditory cortex following medial geniculate TBS is blocked by cortical application of an NMDA receptor antagonist (AP-5; W. Speechley and H.C. Dringenberg, unpublished data), also indicative of a cortical, rather than thalamic, site of LTP induction. Thus, it appears that the mechanisms of LTP induction may not be equal across different cortical sensory fields, a question that clearly deserves attention in future work on neocortical plasticity.
A well-characterized effect of basal forebrain stimulation is the suppression of slow, large-amplitude oscillations and increase in faster beta and gamma activity (activation) in the ECoG by means of cortical ACh release (Casamenti and others 1986; Metherate and others 1992; McLin and others 2002b; Fournier, Materi, and others 2004). Thus, it was surprising to us that there was no significant correlation between the effectiveness of basal forebrain stimulation to produce ECoG activation and to enhance thalamocortical LTP. It is known, however, that cortical ACh release and ECoG activation do not always correlate (Rasmusson and others 1996) and that noncholinergic inputs to the cortex also contribute to ECoG activation (Vanderwolf 1988; Dringenberg and Vanderwolf 1998). The fact that ECoG activation does not necessarily provide a good index of the level of ACh release may account for the lack of correspondence between activation and LTP enhancement noted in the present experiments. Alternatively, the basal forebrain corticopetal system also contains a significant number of GABAergic projections, and it appears that these fibers terminate on inhibitory interneurons in the cortex (Semba 2000; Sarter and Bruno 2002). Thus, a potential disinhibition of thalamocortical synapses, in concert with the critically important ACh release, could also contribute to LTP enhancement without necessarily being related to changes in the level of cortical ECoG activation.
The data presented here may have implications for the regulation of experience-dependent cortical plasticity in more naturalistic situations. Recent investigations have demonstrated a surprising level of plasticity at the earliest stages of processing in the mature central visual system. Effects of monocular deprivation occur in adult animals after the end of the “critical period” for the development of ocular dominance columns (Sawtell and others 2003; Pham and others 2004; Tagawa and others 2005). Further, changes in neuronal discharge rate or field potentials in LGN and V1 are induced by behavioral training such as visual discrimination learning (Gibbs and others 1986; Albrecht and others 1990; Sharma and others 2003; Salazar and others 2004). In adult humans suffering from amblyopia, visual training results in substantial improvements in perceptual functioning such as contrast sensitivity (Polat and others 2004). Thus, both the normally developed and dysfunctional adult visual cortices retain plasticity mechanisms that can be engaged by visual experience and training.
Performance in visual tasks is influenced by attentional processes, thought to depend on cortical cholinergic mechanisms (Muir and others 1994; McGaughy and others 1996, 2002; Sarter and Bruno 2000). For example, cortical ACh release is elevated during conditions of increased visual attentional load (Dalley and others 2001; Arnold and others 2002). Even though these studies assessed ACh in frontal cortical regions, other work has shown that visual stimulation can enhance ACh release selectively in V1 (Fournier, Semba, and Rasmusson 2004). Thus, ACh is in a position to influence V1 plasticity during naturalistic visual processing. The results of the present investigation predict that reorganization of V1 in adult animals should be greatly facilitated under conditions of heightened release of endogenous ACh or pharmacological stimulation of cholinergic transmission.
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada to HCD. Conflict of Interest: None declared.